
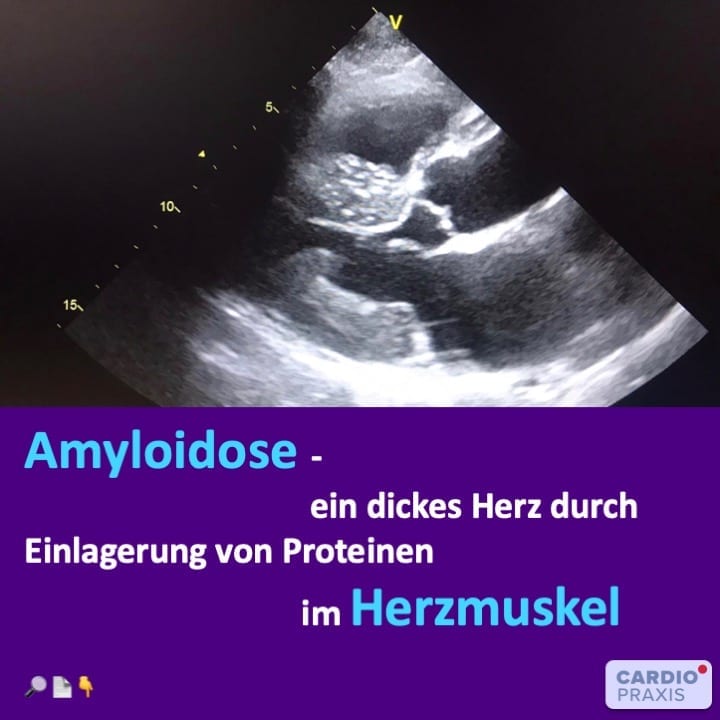
Amyloidosen: überschüssige Eiweißbildung mit Ablagerung in Organen
Amyloidosen sind ein Oberbegriff für verschiedene Eiweißspeicher-Erkrankungen, die sich in Ursachen, Prognosen und Therapien unterscheiden. Hierbei produzieren Zellen unseres Körpers strukturell veränderte Proteine (Eiweiße), die durch die Fehlbildung in Eiweißfäden, sogenannte Amyloidfibrillen, zerfallen. Dabei kann unser Körper die Amyloidfibrillen nicht abbauen oder ausscheiden, so dass es zu einer Ablagerung überschüssiger Proteine in verschiedenen Organen kommt. Diese Amyloidfibrillen lagern sich nicht nur im Herzgewebe ab, sondern auch in Niere, Magen-Darm, im peripheren und autonomen Nervensystem, in Leber, Weichteilen und Gelenken. Infolge kann es durch die überschüssige Eiweißablagerung zur Fehlfunktion und schlimmstenfalls zum Organversagen kommen.
Kardiale Amyloidose: AL-Amyloidose und ATTR-Amyloidose betreffen am häufigsten das Herz
Bei der AL-Amyloidose besteht in ca. 50-70% der Fälle eine Herzbeteiligung, ansonsten ist häufig die Niere betroffen. Dabei beträgt das Auftreten der AL-Amyloidose in Nordamerika ca. 1:100.000 pro Jahr. Für Deutschland liegen allerdings noch keine exakten Zahlen vor, da die Erkrankung erst seit 2018 in einem Register erfasst wird. Die Krankheitsursache ist zudem eine Erkrankung der Plasmazellen, die bei Gesunden Antikörper bilden. Bei der AL-Amyloidose kommt es zu einer pathologischen Vermehrung der Plasmazellen mit überschüssiger Proteinproduktion von Immunglobulin-Leichtketten, die normalerweise auch ein Bestandteil der Antikörper sind. Diese strukturell veränderten Leichtketten werden dann als Amyloid abgelagert. Die Vermehrung von monoklonalen Plasmazellen bezeichnet man auch als monoklonale Gammopathie. Diese selbst ist noch nicht bösartig und bedarf oft nur einer Beobachtung. Sie kann aber auch eine Vorstufe für eine bösartige hämatologische Erkrankung (multiples Myelom, Lymphom) sein oder auf eine zugrunde liegende Amyloidose hinweisen.
Die Transthyretin-bedingte Amyloidose betrifft hauptsächlich das Herz und die peripheren Nerven. Dabei unterscheiden wir zwei Unterformen: die angeborene (hereditäre) und die erworbene ATTR-Amyloidose. Die angeborene ATTR-Amyloidose ist Folge einer Genmutation und tritt sehr selten auf (1:100.000 pro Jahr). Die altersbedingte, nicht erbliche Form der ATTR- Amyloidose wird oft Wildtyp (wtATTR) genannt und tritt häufiger auf. Sie betrifft hauptsächlich ältere Männer im Alter von über 65 Jahren. Neuere Studien weisen zudem darauf hin, dass die wtATTR oft unterdiagnostiziert ist. So konnten in einer Autopsiestudie bei 19% der Patienten mit einer diastolischen Herzinsuffizienz (HFpEF) Amyloidablagerungen nachgewiesen werden. Auch bei verkalkter Aortenklappenstenose wiesen 32% der über 74-jährigen Männer Amyloidablagerungen auf. Der Auslöser der Erkrankung liegt bei beiden Formen in der fehlerhaften Produktion des Transport-Proteins Transthyretin in den Leberzellen. Transthyretin übernimmt normalerweise eine Transportfunktion für Vitamin A und Schilddrüsenhormone. Bei Patienten mit ATTR-Amyloidose fällt dieses Transportprotein auseinander, so dass sich Amyloidfibrillen im Herzgewebe ablagern.
Herzbeteiligung bei Amyloidose prognosebestimmend
Die Prognose der kardialen Amyloidose ist schlecht. Eine wesentliche Ursache dafür liegt in der oft späten Diagnosestellung, durchschnittlich nach 2 Jahren. Dadurch ist die Sterblichkeit sehr hoch. Sterblichkeitsraten von 40% innerhalb von 2 Jahren werden dabei angegeben. Besonders die Wildtyp-ATTR-Amyloidose (wtATTR) infiltriert alle Strukturen des Herzens: neben der Herzwand auch das Reizleitungssystem, die Herzklappen und Herzkranzgefäße. Daher sehen wir bei der wtATTR-Amyloidose häufiger eine stärkere Wandverdickung des Herzens als bei anderen Amyloidoseformen.
Fazit:
Die wesentlichen Formen der kardialen Amyloidose sind die AL- und die ATTR-Amyloidose, die unterschiedliche Ursachen haben. Die Prognose der kardialen Amyloidose ist schlecht, eine frühzeitige Diagnose ist daher wichtig.






