

Die Leber verstoffwechselt Medikamente
Damit Medikamente optimal wirken und zu keinen Nebenwirkungen führen, muss der menschliche Körper sie verstoffwechseln können. Manchmal ist es erst die biochemische Verarbeitung durch den Körper selbst, die aus einer sogenannten Pro-Drug, einem „Vormedikament“, die eigentlich wirksame Substanz „herstellt“.
Besonders wichtig für die Verstoffwechselung von Medikamenten ist die Leber. Sie ist das erste große Stoffwechselorgan, welches dem Darm und somit dem Aufnahmeort der meisten Präparate, nachgeschaltet ist. In der Leber wiederum ist es auf zellulärer Ebene das Cytochrom-P450-System, das vor allen anderen Systemen für den Ab- und Umbau von Medikamenten am wichtigsten ist, denn 90% aller Medikamente werden hier verarbeitet.
Cytochrome, auch CYPs genannt, sind Eiweiße, die vor allen Dingen wasserunlösliche Substanzen in wasserlösliche Stoffe umwandeln. Dadurch können diese besser ausgeschieden werden, zum Beispiel über die Niere. Für unterschiedliche Medikamente benötigt der Organismus unterschiedliche CYPs. So hat der Mensch 57 verschiedene CYPs. Eines der wichtigsten Cytochrome ist CYP3A4, hierüber werden bis zu 50 Prozent aller Medikamente verarbeitet.
Stoffwechsel von Medikamenten: bei jedem Menschen unterschiedlich durch Genetik
Menschen sind auch in Bezug auf das Cytochrom-System genetisch unterschiedlich. Variiert ein einzelnes CYP, z.B. CYP3A4 in seiner Ausprägung, dann nennen wir das einen Genpolymorphismus. Diese Genpolymorphismen sind ein wesentlicher Grund dafür, dass Menschen auf Medikamente mit unterschiedlicher therapeutischer Wirkung und mit unerwünschten Nebenwirkungen reagieren.
Wir unterscheiden:
- slow metabolizer
- normal metabolizer
- fast metabolizer
„Slow metabolizer“ bedeutet, dass ein CYP bedingt durch eine genetische Veränderung vergleichsweise langsam arbeitet. „Fast metabolizer“ steht dafür, dass ein CYP besonders schnell eine angebotene Substanz umwandelt. Das erklärt auch, warum eine Substanz bei einigen Menschen gut wirken kann („normal metabolizer“), bei anderen aber nicht so gut, weil es z.B. zu schnell abgebaut wird („fast metabolizer“).
Über genetisch festgelegte Veränderungen erklärt sich auch ein Teil der Nebenwirkungen von Medikamenten. Ein zu langsamer Abbau des medizinischen Wirkstoffes („slow metabolizer“) kann eine überstarke Anhäufung des Medikaments im Körper zur Folge haben. Das wiederum verursacht toxische Nebenwirkungen und der Körper ist überfordert.
Medikamente und Nahrungsmittel beeinflussen CYPs des Cytochrom P450 Systems
Medikamente und Nahrungsmittel können die Funktion von CYPs beeinflussen. Wir unterscheiden:
- Substrate
- Inhibitoren
- Induktoren
„Substrat“ bedeutet, dass ein Medikament von einem bestimmten CYP verarbeitet wird, z.B. Metoprolol über CYP 3A4. Der Stoffwechsel hängt also hier wesentlich davon ab, wie ein einzelnes CYP genetisch festgelegt ist.
Problematisch wird es bei „Inhibitoren“. Sie blockieren mehr oder weniger ein CYP und hemmen so den Abbau von anderen Medikamenten. Das können übrigens auch Nahrungsmittel sein, wie z.B. Grapefruit bei CYP3A4.
„Induktoren“ hingegen beschleunigen die Stoffwechselrate von CYPs, so z.B. Johanniskraut bei CYP3A4. Entsprechende Medikamente werden dann schneller abgebaut und sind bei den betroffenen Menschen weniger wirksam. Das gilt übrigens auch für die kombinierte Einnahme der Anti-Baby-Pille mit Johanniskraut.
Ein Medikament beeinflusst häufig mehrere Cytochrome
Die meisten Menschen bekommen gleich mehrere Pillen verschrieben, z.B. bei der Behandlung von Koronarer Herzkrankheit, Herzschwäche oder Bluthochdruck. So wird der Abbau von Medikamenten noch komplizierter.
Verschiedene Medikamente werden über denselben Stoffwechselweg abgebaut, z.B. Metoprolol (Beta-Blocker), Lercanidipin (Kalziumantagonist) und Irbesartan (AT-Blocker) über CYP3A4. So kann auch bei einem CYP3A4 „normal-metabolizer“ zu einer Überlastung von CYP 3A4 auftreten. Erstrecht, wenn es sich bei dem behandelten Menschen um einen „slow-metabolizer“ handelt.
Meistens nutzt dasselbe Medikament verschiedene CYPs für den Abbau. Der häufig eingesetzte Beta-Blocker Metoprolol z.B. wird über CYP3A4 und CYP2D abgebaut. Andere Substanzen nutzen weit mehr und bis zu 7 CYPs. Bei den häufig genutzten Medikamenten des Herz-Kreislaufbereichs interagiert ein Medikament (Substrat, Inhibitor, Induktor) mit durchschnittlich mit 2,1 CYPs.
Genvarianten des Cytochrom-System bestimmen: ein Schritt in die richtige Richtung
Genetische Veränderungen von Cytochromen kann man heutzutage unkompliziert und zuverlässig molekular-genetisch mit Hilfe einer Blutprobe im Labor bestimmen. Allerdings liegen die Kosten für die notwendige komplette Charakterisierung zwischen 300 und 400 Euro. Diese werden nur von einem Teil der privaten Krankenkassen und gar nicht von den gesetzlichen Krankenkassen übernommen.
In der Cardiopraxis empfehlen wir Ihnen einen solchen Test, falls aufgrund von medikamentösen Nebenwirkungen der begründete Verdacht auf einen sogenannten Genpolymorphismus besteht. Wir raten Ihnen vor allen Dingen dann dazu, wenn Sie mit einer lebenslangen Einnahme von Medikamenten rechnen müssen. So können die aktuellen Medikamente überprüft werden und auch bei neuen Präparaten später Nebenwirkungen vermieden werden.
Medikamenteninteraktionen: So gehen wir praktisch in der Cardiopraxis vor
Es folgen jetzt einige Abschnitte, die eigentlich für Ärztinnen und Ärzte gedacht sind, die lernen wollen mit dem Cytochrom-System praktisch umzugehen. Falls Ihnen das verständlicherweise zu kompliziert ist, dann können Sie gerne zum letzten Abschnitt „Nebenwirkung von Medikamenten…“ springen.
Leider stehen uns molekular-genetische Analysen nur gelegentlich zur Verfügung. Wie gehen wir da in der Cardiopraxis nun praktisch vor? Wir führen eine Cytochrom-Liste mit fast allen Medikamenten, die wir zur Therapie Ihres Herz-Kreislaufsystems nutzen. In dieser Liste ordnen wir den Medikamenten die verschiedenen CYPs mit ihren jeweiligen Eigenschaften zu. Diese Liste ist nur ein Beispiel wie Sie die Zuordnung machen können.1
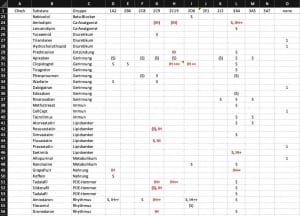
Schritt 1: Verdacht schöpfen
Alles beginnt mit einem Problem. Bei zahlreichen Medikamenten kennen wir das Potenzial für Interaktionen. Z.B. ist das Herzrhythmusmedikament Amiodaron ein starker Inhibitor von CYP2C9 und Blutdrucksenker wie Irbesartan und Candesartan müssen in der Regel deutlich geringer dosiert werden.
Wenn ein Mensch von einer neuen Nebenwirkung berichtet, dann kann das schon auf eine Abbaustörung durch ein verändertes CYP hinweisen. Das gilt vor allen Dingen dann, wenn es sich um allgemeine Unverträglichkeit handelt.
Ein weiterer Ansatzpunkt: Wenn bei einem neuen Medikament eine stärkere Wirkung einer schon länger genommenen Substanz eintritt. Stellen Sie sich zum Beispiel vor, dass Sie seit Jahren den Beta-Blocker Metoprolol in niedriger Dosierung einnehmen. Dabei lag Ihre Herzfrequenz in Ruhe bisher immer bei ca. 72 bpm. Nun erhalten Sie im Krankenhaus zusätzlich den Thrombozytenblocker Clopidogrel. Sie berichten nun über neu aufgetretene Benommenheit und eine Herzfrequenz von 56 bpm.
Was ist passiert?
Schritt 2: Interaktion zwischen Medikament und Cytochrom klären
Im ersten Schritt schauen wir, ob das unter Nebenwirkungsverdacht stehende Clopidogrel überhaupt über ein CYP verarbeitet wird. Wenn wir unsere CYP-Liste nicht zur Hand haben, dann schauen wir im Internet in der Datenbank „drugbank“ nach. Hier finden wir unter „Predicted ADMET Features“ die sogenannte pharmakokinetische Eigenschaft des Medikaments: „A“ steht für „Adsorption“ (= Aufnahme), „D“ für „Distribution (= Verteilung), „M“ für „Metabolism“ (= Stoffwechsel), „E“ für „Excretion“ (= Ausscheidung) und „T“ für „Toxicity“ (= Giftigkeit).
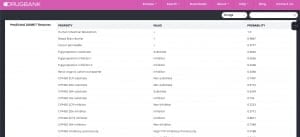
In der Spalte 1 „Property“ sind die potenziellen Eigenschaften des Medikaments in Bezug auf das CYP-System aufgelistet. Darauf wurde es laborchemisch getestet, in diesem Fall die Eigenschaft als CYP2D6 Inhibitor.
In Spalte 2 unter „Value“ ist angegeben, welche Eigenschaft das Medikament tatsächlich hat, z.B. Inhibitor. In der Tat ist Clopidogrel ein Inhibitor von CYP2D6.
Und in Spalte 3 finden Sie die Wahrscheinlichkeit dieser tatsächlichen Eigenschaft. Dabei werten wir 1,0 als hoch und <0,50 als niedrig. Im Fall von Clopidogrel besteht eine mittlere Wahrscheinlichkeit von 0,57, dass Clopidogrel ein Wirkung als „Inhibitor“ hat.
Schritt 3: Schnittmenge mit anderen Medikamenten finden
Nun haben Sie herausgefunden, dass Ihr Medikament Clopidogrel tatsächlich Einfluss auf das Cytochrom-System hat. Sie müssen jetzt sehen, ob andere Medikamente mit Clopidogrel eine Schnittmenge beim CYP2D6 haben. Das können Sie wieder über „drugbank“ machen. Bei mehreren Medikamenten ist es manchmal einfacher, wenn Sie im englischen Wikipedia den Eintrag zum Cytochrom selbst lesen, in diesem Fall „CYP2D6″. Am Ende des Wikipedia-Beitrages finden Sie eine ziemlich vollständige Liste mit Medikamenten, die als Substrate, Inhibitoren oder Induktoren mit dem Cytochrom interagieren können.
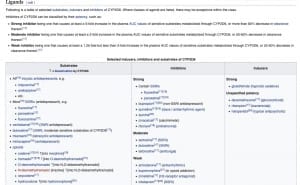
Schritt 4: Konsequenzen ziehen
Falls wir den klinischen Verdacht auf einen Genpolymorphismus haben, schreiben wir das als Verdachtsdiagnose in unsere Arztbriefe. Wir versuchen dann in der Zukunft Medikamente zu vermeiden, die über diesen Stoffwechselweg abgebaut werden.
Nebenwirkungen von Medikamenten – schwierige Analyse für Ärztin und Arzt
Sie werden spätestens jetzt bemerkt haben, dass die Bewertung von Nebenwirkungen sehr komplex, schwierig und zeitaufwändig ist, auch für den verschreibenden Arzt. Das bedeutet: Die Empfehlung von Medikamenten ist folglich auch immer mit einem hohen Maß an Verantwortung verbunden.
Sie verstehen jetzt auch, warum es ab der Einnahme von 5 Medikamenten für eine Ärztin bzw. einen Arzt fast unmöglich ist, die Arzneimittelinteraktionen zu bewerten. Und hinzu kommt, dass auch pflanzliche Präparate, wie zum Beispiel Johanniskraut das Cytochrome-System beeinflussen können.
Das alles erklärt auch unseren Grundsatz in der Cardiopraxis: „So wenig Medikamente wie nötig“. Es geht hierbei nicht darum, Ihren individuellen Wünschen nach wenigen Präparaten zu folgen, sondern einfach um eine medizinische Notwendigkeit.
Wir rechnen in der Zukunft damit, dass die Bestimmung von Arzneimittelinteraktionen mit Medikamenten besser bestimmbar und besser vorhersehbar werden. Die bisherig eingesetzten einfachen Ampelsysteme, z.B. durch Apotheken sind noch zu grob. Ziel muss sein, die individuelle genetisch determinierte Stoffwechselaktivität von Cytochromen bzw. ihre Aktivität bei Einsatz mehrerer Medikamente zu erfassen. Hilfreich werden hierbei System der künstlichen Intelligenz sein, die große Datenmengen verarbeiten können. Optimal ist eine molekular-genetische Analyse des Cytochrom-Systems bei allen Menschen, die langfristig Medikamente einnehmen.
Anhang
Links zu einzelnen Medikamenten
Geben Sie oben einen Wirkstoff ein und scrollen Sie nach unten bis Sie zum Abschnitt „Properties“ gelangen. Dort finden Sie unter „Predicted ADMET Features“ die Bedeutung der Medikamente im Verhältnis zu verschiedenen CYPs.
Links zu einzelnen Cytochromen
Wikipedia-Links zu den wichtigsten CYPs für Herz-Kreislaufmedikamente.
| 1A2 | 2B6 | 2C8 | 2C9 | 2C19 | 2D6 | 2E1 | 2J2 | 3A4 | 3A5 |
Rot markierte Links kennzeichnen Beiträge, die Medikamentenlisten enthalten; Sie finden sie dort unter „Ligands“.
1Für die CYP-Liste übernimmt Cardiopraxis keine Gewährleistung. Nur zum internen Gebrauch in der Cardiopraxis gedacht.
Cardiopraxis – Kardiologen in Düsseldorf & Meerbusch





